New publication in 2022: Computational Material Science (IF: 3.572 )
Abstract
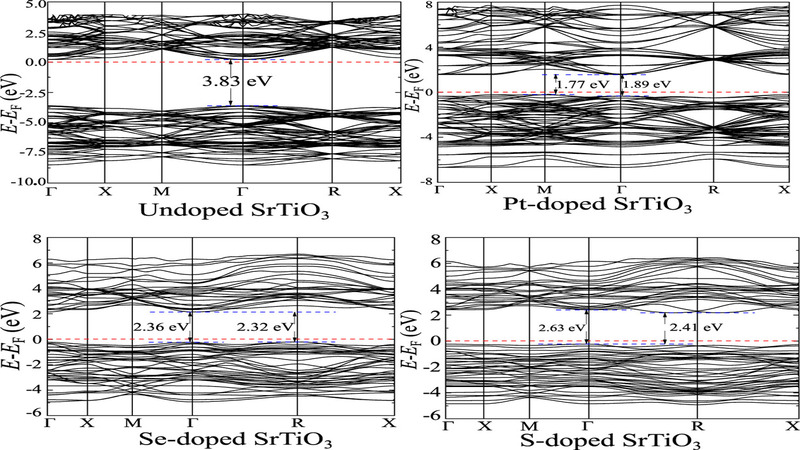
We present a Hubbard U optimized density functional theory based study of band gap engineered doped SrTiO3 suitable for light sensitive applications. We tune the Hubbard U parameter Uopt to match the experimental direct band gap of SrTiO3. We benchmarked the DFT+Uopt derived density of states, band structure and optical properties with that of sophisticated Heyd–Scuseria–Ernzerhof (HSE06) hybrid functional simulations. The reliability of the DFT+U method is further tested in simulating stability of the cubic SrTiO3 from elastic tensor and density functional perturbation theory based phonon band structure calculations. Moreover, the DFT+Uopt simulated SrTiO3 are identified against the experimental observations found in existing literature. We red shift the of SrTiO3 within the DFT+Uopt formalism from ultraviolet to visible range by incorporating different such as Pt, S and Se which is consistent with recent HSE06 hybrid functional based simulations found elsewhere. The optical absorption simulations revealed steep of doped SrTiO3 in the visible spectrum. Overall, the DFT+U approach successfully probed the potential of doped SrTiO3 in solar harvesting applications.
Link: